Integration Methods for Molecular Dynamics Approach
The step by step solution of the equations of motion using a finite difference approach is performed by the use of an integration algorithm in molecular dynamics simulations. One common algorithm is the Verlet algorithm. This is derived from a Taylor expansion of the positions about time t as below equations:
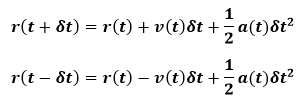
from these equations, we can write:
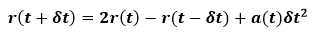
As can be seen, the Verlet algorithm is time-reversible. Also the velocities are absent from final equation but can be calculated from below equation:
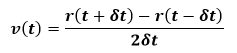
This is necessary for the calculation of the kinetic energy. The Verlet algorithm is simple and compact to code and the time-reversal symmetry leads to good energy conservation. However, the velocities are not well handled. Also r equation involves the addition of a small number onto a large number, which can introduce numerical inaccuracies. Two alternative formulations of the Verlet algorithm have been proposed to remedy this shortcoming. The first is the leapfrog algorithm as below:
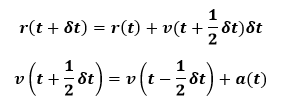
Here the mid-step velocities are calculated using final equation and then these are using to calculate the new positions. The velocity at time t may then be calculated from below formalism:
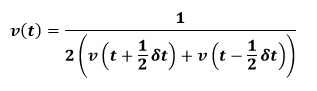
Another alternative is the Velocity-Verlet algorithm. This takes the form:
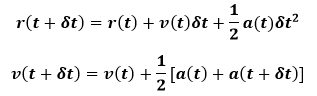
This is implemented in two stages. First the positions are advanced. The mid-step velocities are then calculated from the acceleration at time t by below equation:
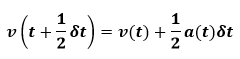
The accelerations at final time are then calculated and the velocity move is completed as below:
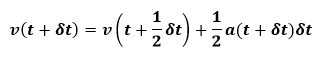
Reference
Verlet, L. (1967). Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Physical Review, 159(1), 98–103.
doi:10.1103/physrev.159.98.