Consistent Valence Force Field (CVFF)
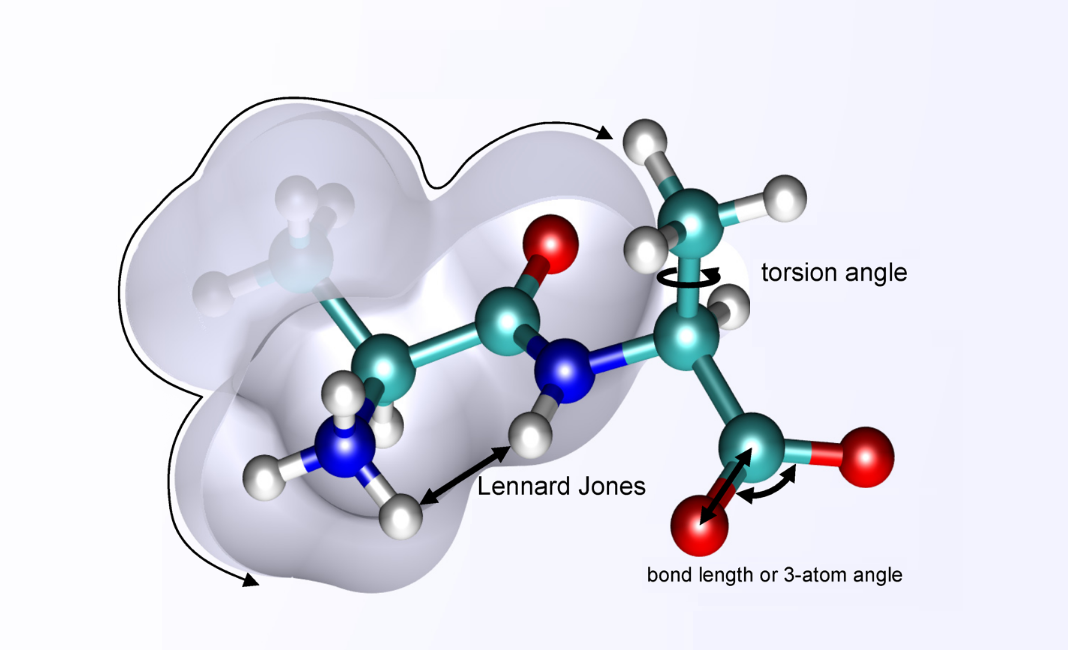
The interatomic potential is an important parameter in molecular dynamics simulations. The Consistent Valence Force Field (CVFF) is one of the important force-fields used in the atomic description of various systems. CVFF, the original force-field provided with the Discover program, is a generalized valence force-field. Parameters are provided for amino acids, water, and a variety of other functional groups. Theoretically, the CVFF force-field is formulated as below,
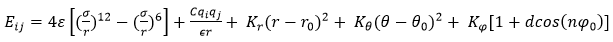
where, r is the distance between two interacting atoms, ε is the depth of the potential well, and σ is the distance at which the atom-atom potential energy is zero. In the Coulombic equation (second component of the defined equation) C parameter is an energy-conversion constant, qi and qj are the charges on the 2 atoms, and ϵ is the dielectric constant. In the harmonic component, Kr/Kθ/Kφ defines harmonic constant and r0/θ0/φ0 represents equilibrium distance/angle constant. Also, the improper component defined by the below formalism,
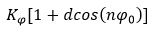
Each of described constants chooses from CVFF reference in molecular dynamics simulations.

Technically, the CVFF force-field can be implemented in LAMMPS software. For the various interaction settings (bonded and non-bonded interactions) in the “LAMMPS script” below commands should be used:
pair_style lj/cut/coul/long
pair_coeff …
bond_style harmonic
bond_coeff …
angle_style harmonic
angle_coeff …
dihedral_style harmonic
dihedral_coeff …
improper_style cvff
improper_coeff …
the “…” in these commands are coefficient for each type of interactions which set for all atom types in modeled samples from CVFF reference.
Reference
[1] Pnina Dauber-Osguthorpe; Victoria A. Roberts; David J. Osguthorpe; Jon Wolff; Moniqe Genest; Arnold T. Hagler (1988). Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase-trimethoprim, a drug-receptor system. 4(1), 31–47. doi:10.1002/prot.340040106.